General options:
For most functions, you will get a specific window for setting
parameters and options. Some options are identical for most of
the functions:
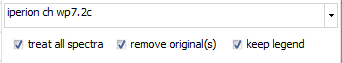
- At the top, there is a dropdown selector containing the current
spectra list. Here you can select the spectrum to be processed
(if only one spectrum gets treated). If a preview is available,
the spectrum is selected here.
- if the "treat all spectra" option is checked,
all spectra will get processed and attached as new spectra at
the end of the spectrum list. Otherwise, only the currently selected
spectrum gets processed.
- With the "remove all" option checked, the original
spectra will be removed and only the processed spectra/ spectrum
remain(s) .
- The "keep legend" option leaves the original
legend text unchanged. If unchecked, a function-specific text
part will be added to the legend text.
|
|

|
Basic
arithmetics
Add  will add the y values of the two spectra selected in the dropdown
lists. If "treat all spectra" is selected, the spectrum
selected in the lower list will be added to all spectra.
will add the y values of the two spectra selected in the dropdown
lists. If "treat all spectra" is selected, the spectrum
selected in the lower list will be added to all spectra.
Subtract
 will subtract the y values of the lower spectrum from the upper
spectrum. If "treat all spectra" is selected, the spectrum
selected in the lower list will be subtracted from all spectra.
will subtract the y values of the lower spectrum from the upper
spectrum. If "treat all spectra" is selected, the spectrum
selected in the lower list will be subtracted from all spectra.
Multiply
 will multiply the y values of the two spectra selected in the
dropdown lists. If "treat all spectra" is selected,
the spectrum selected in the lower list will be multiplied with
all spectra.
will multiply the y values of the two spectra selected in the
dropdown lists. If "treat all spectra" is selected,
the spectrum selected in the lower list will be multiplied with
all spectra.
Divide
 will divide the y values of the upper spectra from the lower spectrum.
If "treat all spectra" is selected, all spectra will
get divided by the spectrum selected in the lower list.
will divide the y values of the upper spectra from the lower spectrum.
If "treat all spectra" is selected, all spectra will
get divided by the spectrum selected in the lower list.
|
| <jump
back to top> |
|
|
Averaging
The y values
of the selected spectra (left mouse) will be averaged, using either
arithmetic mean or median as calculation method. For selecting
spectra to be averaged, some sophisticated options are available
for spectra series, based on naming or numbering schemes within
the legend texts. The legend text of the averaged spectrum can
be entered manually, or derived from one of the used spectra by
left-mouse double-clicking its entry.
Merging
The selected
spectra (left mouse) will be merged into a single spectrum, by
averaging the overlapping parts with smooth transitions, or interpolating
gaps. For selecting spectra to be merged, some sophisticated options
are available for spectra series, based on naming schemes within
the legend texts, or automated assignment for spectra coming as
sub files from a common spectrum file( e.g. from Avantes multi-channel
system). The legend text of the merged spectrum can be entered
manually, or derived from one of the used spectra by left-mouse
double-clicking its entry.
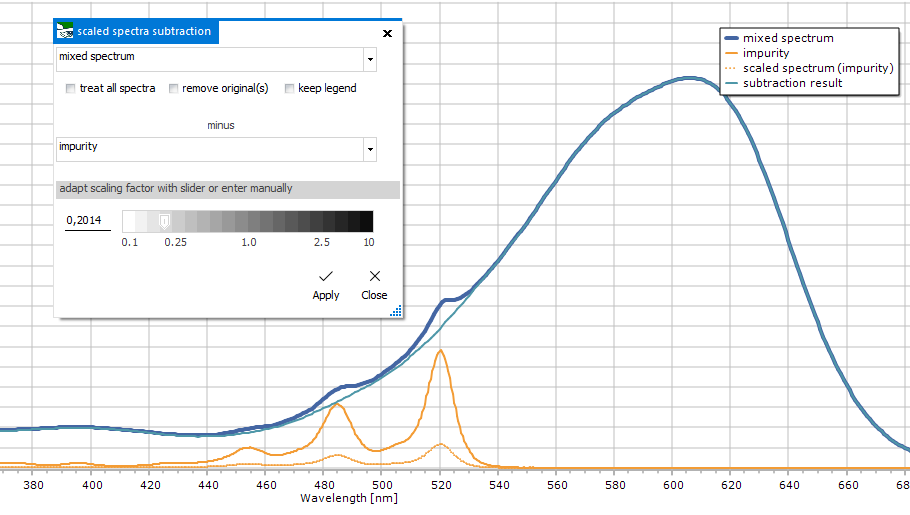
Scaled Subtraction

For subtracting
a spectrum after scaling it with a factor. This factor can be
determined interactively by moving the slider or manually entering
the scaling factor. During this, the plot will show and live update
the scaled spectrum and the subtraction result.
 |
| <jump
back to top> |
|

|
Transmittance
& Reflectance (from Intensity)
From two intensity
spectra (sample and background/ reference), the transmittance
or reflectance will be calculated after T
= I / I0, resp. R
= I / I0
- For transmittance, the reference spectrum needs to be
measured from full light source intensity, and the sample spectrum
by passing this light through the sample.
- For reflectance, the reference spectrum needs to be measured
from full light source intensity reflected from a white reflectance
standard, and the sample spectrum by measuring this light source
reflected from the sample.
Absorbance
(from Intensity)
From two intensity
spectra (sample and background/ reference), the absorbance will
be calculated after
A = -log(T)
with T = I / I0
The reference spectrum needs to be measured from full light
source intensity, and the sample spectrum by passing this light
through the sample.
|
| <jump
back to top> |
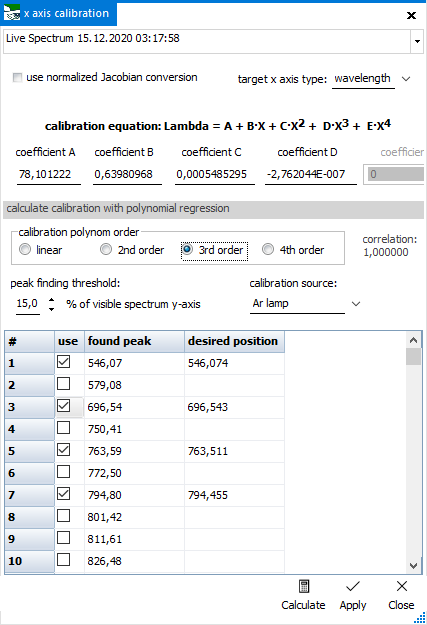 |
X Axis Pixel
Calibration
For spectra
with uncalibrated x axis (pixels), or when you need to recalibrate.
Mostly used for measured spectra from controlled spectrometers,
therefore also available from the Acquire ribbon. The calibration
procedure consists of two steps:
1) calculating the calibration parameters (linear or 2nd to 4th
order polynomial), based on a spectrum with known peak positions.
Those peak positions get mapped onto the known positions with
a polynomial regression.
2) applying the polynomial calibration parameters upon the selected
spectrum's x axis values and assigning the new axis type.
It is possible
to calibrate for
- absorbance: by using a sample with known peaks, like
a Holmium or Didymium filter. Target: wavelength scale
- fluorescence: by using a light source with known emission
peaks, like a pen lamp source (Hg, Hg/Ar, Xe, Ne). Target: wavelength
scale
- Raman shift: by using a sample with known Raman lines,
as described in ASTM E1840 standard guide. Target: Raman shift
scale
For step 1),
, open
the calibration dialog from the button's dropdown menu. In the
upper field, select the spectrum to be used. Change the peak finding
threshold, if necessary. Select the desired x axis type after
calibration on the right side. Below, the calibration coefficients
will be shown, after doing the polynomial regression.
In the table below, the x axis positions of the found peaks from
the selected spectrum are displayed. You can either enter the
known positions into the "desired position" column manually,
or select from a number of precompiled positions that are shown
as dropdown list. The content of this dropdown list depends on
the calibration source chosen above. Only the "custom"
option allows to manually enter position values. Check the values
to be used during calculation.
Finally, select the polynomial order for the regression calculation
and press the "Calculate" button. The calculated
calibration coefficients will show up in the respective fields
above, together withe the regression's correlation coefficient.
To keep the coefficients
and apply on the selected spectrum (step 2), click the "Apply"
button.
The peak data
for the selectable calibration sources is located in a file called
"calibration_lines.csv" in the program folder, this
file's content can be changed to adapt or enhance the range of
available calibration peak data.
The calibration
parameters will be remembered for the current session. To make
them a permanent setting, go to File > Options > Transform
> calibrate x axis
For the calibration
creating Raman spectra from wavelength spectra, the area-preserving
calculation option "use normalized Jacobian conversion"
is available. This takes care of the distortion of intensity values
caused by the inverse relationship between wavelength and energy,
as explained in this publication
and its correction.
HINT: To better
find the peaks to be used during calibration, you might turn on
"Peak labels" before starting the calibration procedure.
|
| |
<jump
back to top> |
|

|
Raman Shift
Usually, Raman
spectra get measured as wavelength spectra, and are then transformed
into Raman spectra, having Raman Shift als x axis type (shown
as wavenumbers), calculated after this equation: RamanShift
= 10^7/Lambda_exc - 10^7/Lambda
The RamanShift is a relative parameter, based on wavelength distance
to the actual excitation laser wavelength. therefore, the laser
wavelength is always required as input value. It is recommend
to use the accurate value, just entering "785" for a
785nm Raman spectrometer wont do the trick, you need the accurate,
actual value! Otherwise, you might be several wavenumbers off
the truth...
As the spectrum's Rayleigh area near the zero point of the Raman
Shift scale is often highly distorted, it can be omitted for the
transformation, by selecting "cut off Rayleigh peak area"
and entering a cutoff value into the "begin Raman spectrum
at:" field.
For the transformation
from wavelength spectra to Raman spectra, the area-preserving
calculation option "use normalized Jacobian conversion"
is available. This takes care of the distortion of intensity values
caused by the inverse relationship between wavelength and energy,
as explained in this publication
and its correction.
|
| <jump
back to top> |
|

|
Converting
reflectance spectra into "absorbance"
Contrary to
transmittance spectra, there is no easy way to obtain a corresponding
absorbance spectrum from reflectance spectra. Two different methods
are available, as explained below. A third one would be based
on scattering theory using Kramers-Kronig relations, but this
is currently not available in Spectragryph.
Log(1/R) method
The used equation
is already in the name, and looks liike the analogue to caculation
fo absorbance from transmittance. The resulting spectra is shown
with "log(1/R)" as y unit. This is sometimes
used as quick way for creating something absorbance-like, and
seems to work best on totally opaque samples. Much discussed
on the internet...
Kubelka-Munk
transformation
For the calculation
of absorbance from diffuse reflectance spectra, best used on samples
with semi-infinite layer thickness, after this equation: k/s
= (1-R)^2 / (2*R)
The resulting spectra is shown with "Kubelka-Munk
units (K/S)" as y unit.
For a more detailed treatment of both methods, also refer to the
Wiki entries on Kubelka-Munk
theory and Diffuse
reflectance spectroscopy
|
| <jump
back to top> |
|
|
Derivative
The first, second,
third or fourth derivative of the spectrum selected in the drop
down list will be calculated. The smoothing option is strongly
recommended for all derivatives higher than the first derivative.
Interpolate,
Resample
This is the
only place to assign a differing x or y axis type to existing
data (use with care!). It is also possible to redefine start value,
end value and x step width for interpolation to a new set of data
points. This can be useful for bringing diverse spectra onto a
common scale, before further processing with a chemometric package.
Spectrum from
Image
For the transformation
of a spectrum image file into actual spectral data, by converting
a horizontal cross-section of a loaded image into intensity data.
first load an image file with the "Load image"
button (bmp, gif, jpg and png files accepted), then select the
actual spectrum area with the ROI rectangle (visible on checking
"use spectrum ROI"). For a coarse linear x axis
calibration, you can directly set the start and end value in wavenumbers,
alternatively the normal x axis calibration is also applicable
(see above). The clipping indicator will warn of clipping in any
of the colour channels, which means you are loosing linear correlation
of actual to measured intesity to some extent.
|
| <jump
back to top> |
|
![[SpectraGryph]](gryphon_white_green_96.png "back to SpectraGryph main page")